029 - Heterozygous NPHS1 variants with uncertain significance in a newborn with Finnish-type congenital nephrotic syndrome: A case report and literature review
Sunday, April 27, 2025
8:30am - 10:45am HST
Publication Number: 29.5462
Farzina Zafar, Texas Tech University Health Sciences Center School of Medicine, Amarillo, TX, United States; Mohammed A.. Al-Obaide, Department of Pediatrics, Texas Tech University Health Sciences Center, Amarillo, TX, United States, Amarillo, TX, United States; Tetyana L. Vasylyeva, Texas Tech University Health Sciences Center School of Medicine, Amarillo, TX, United States
PGY3 Texas Tech University Health Sciences Center School of Medicine Amarillo, Texas, United States
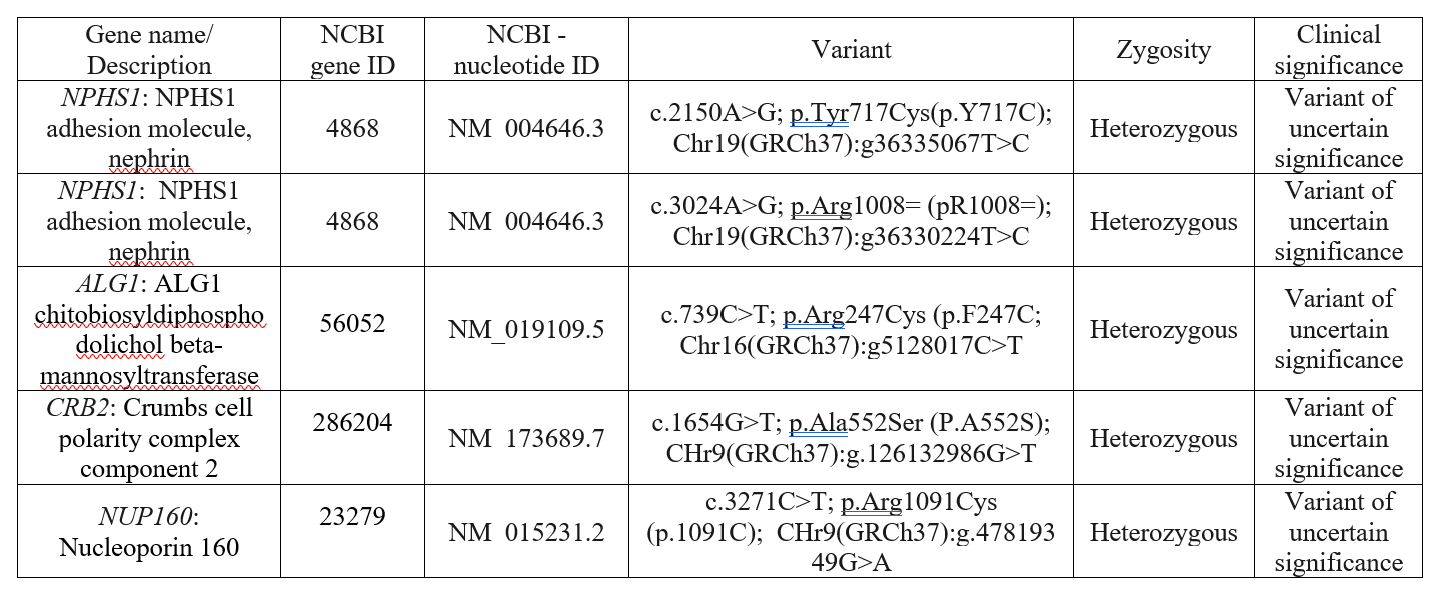
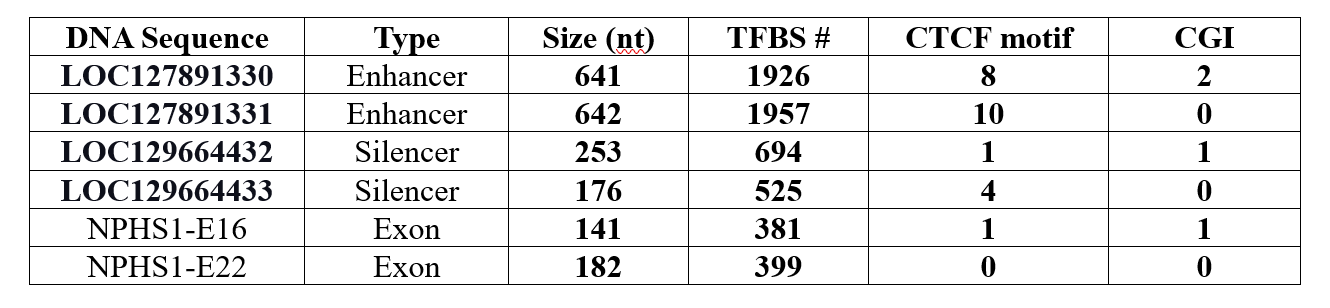
Background: Congenital Nephrotic Syndrome (CNS) is a rare form of nephrotic syndrome that presents within the first three months of life, most commonly as the Finnish type (FCNS). FCNS is an autosomal recessive condition involving mutations in the NPHS1 gene, which encodes nephrin, a crucial protein in kidney filtration. It is characterized by severe proteinuria (excess protein in the urine), hypoalbuminemia (low protein in the blood), and edema (significant swelling), with histological changes like microcystic radial dilatation of the proximal tubules typically appearing after three months. This case report discusses a newborn female with FCNS, who has two heterozygous NPHS1 variants of uncertain significance (VUS) in exons 16 and 22. While VUS has been observed in various conditions like FCNS, Fabry disease, Lynch syndrome, and cancer, their association with the disease remains unclear. This uncertainty highlights the need for further research to understand the pathogenic potential of VUS in FCNS. Objective: To find the relationship between NPHS1 VUS and FCNS. Design/Methods: Whole blood from the infant was analyzed by Next-Generation Sequencing at the Mayo Clinic, testing 56 genes linked to nephrotic syndrome for variants in coding regions. The NPHS1 gene and its mutations, exons, enhancers, and silencers were evaluated using NCBI Gene, NCBI Nucleotide, NCBI Protein, and UCSC Genome Browser databases. Results: A newly identified NPHS1 heterozygous missense variant on exon 16 may affect nephrin protein function. NPHS1 in exons 16 and 22 are potential enhancers due to CpG islands (CGIs), CCCTC-binding factors (CTCF) motifs, and transcription factor binding sites (TFBS), typical of enhancer regions. Disruption of these enhancers could impair gene regulation. Additionally, this VUS may alter exonic splicing enhancers (ESEs), affecting RNA splicing and leading to dysfunctional nephrin, a key factor in gene expression and disease.
Conclusion(s): In conclusion, the link between NPHS1 variants of uncertain significance in exons 16 and 22 and FCNS remains unclear. This study suggests that the p.Tyr717Cys amino acid change from NPHS1 c.2150A>G in exon 16 (Table 1) may impair nephrin function, impacting the Ig strand F of the protein (Figure 1), essential for the kidney filtration barrier. Exons 16 and 22 also harbor regulatory elements, such as ESEs, CGIs, TFBS clusters, and CTCF motifs (Table 2), which may affect nephrin gene expression and contribute to disease. These findings will encourage further experimental research on NHPS1 regulatory elements in the CNS.
The results of the examination of a panel of 52 genes that showed four genes and nucleotide IDs for variants by DNA sequencing in the congenital nephrotic syndrome case study
The exon 16 variant (c.2150A>G; p. Tyr717Cys) may alter the function of Nephrin Ig strand F.
The NPHS1 exons 16 and 22, enhancers, and silencers features and regulatory criteria.

 photo")