
Nephrology 3
Session: Nephrology 3
 photo")
Michael T. Hsieh (he/him/his)
Medical Student
University of Central Florida College of Medicine
Orlando, Florida, United States
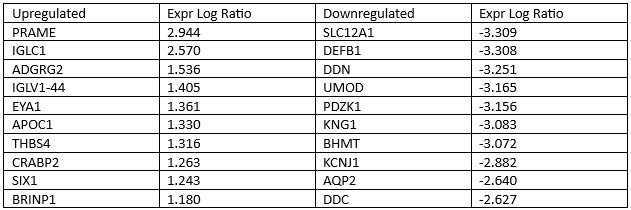 Table 1: Top 10 up and downregulated genes in Wilms tumor compared to healthy controls
Table 1: Top 10 up and downregulated genes in Wilms tumor compared to healthy controls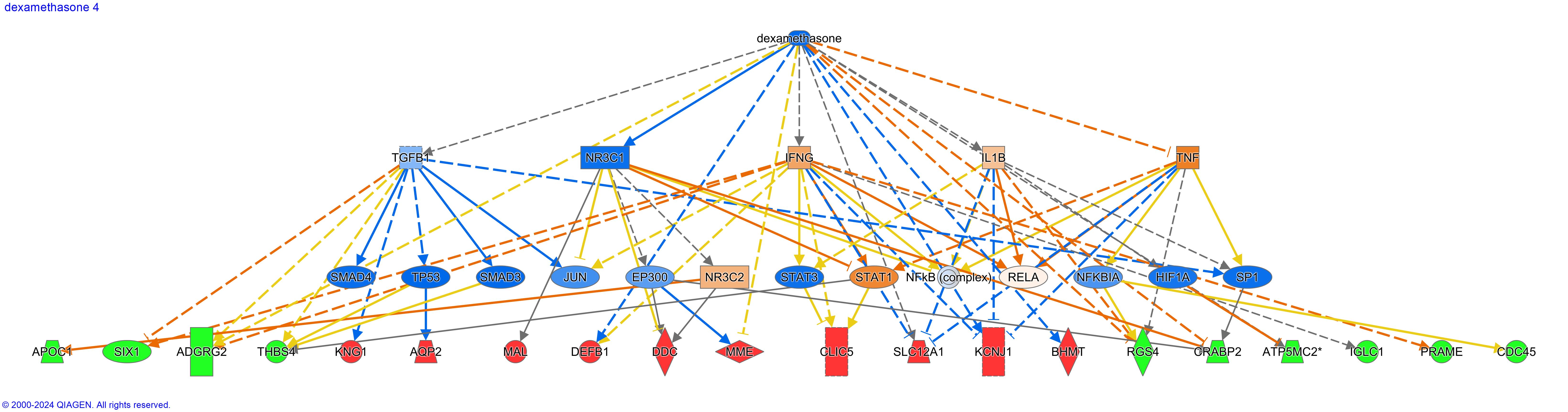 Figure 1. Dexamethasone, top upstream regulator predicted as inhibited with z-score -6.944, and downstream effects of inhibition of dexamethasone.
Figure 1. Dexamethasone, top upstream regulator predicted as inhibited with z-score -6.944, and downstream effects of inhibition of dexamethasone.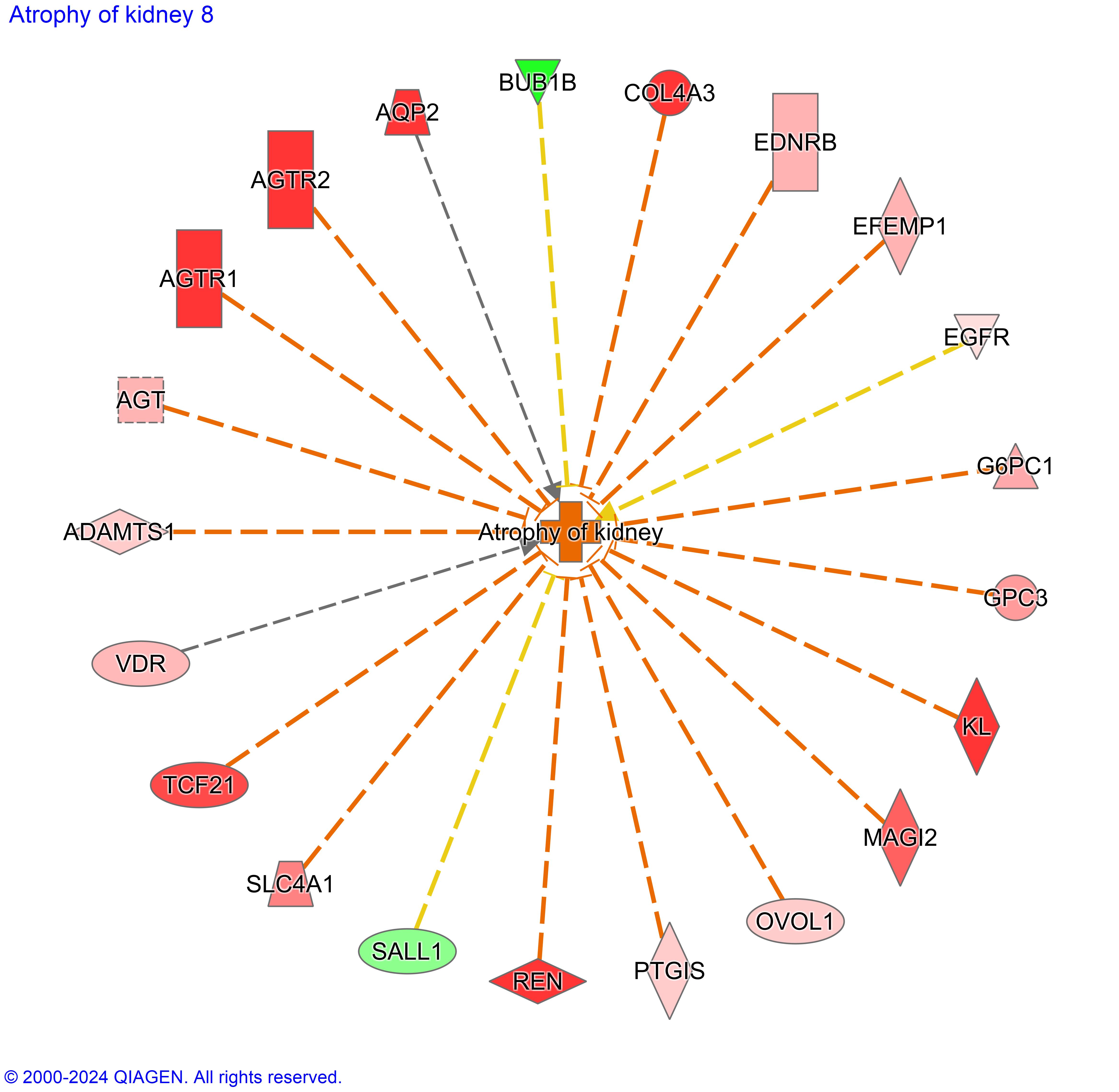 Figure 2. Atrophy of kidney, a top toxicity function associated with Wilms tumor. Atrophy of the kidney has predicted activation with a z-score of 3.146, suggesting the pattern of DEGs in WT contribute to renal atrophy.
Figure 2. Atrophy of kidney, a top toxicity function associated with Wilms tumor. Atrophy of the kidney has predicted activation with a z-score of 3.146, suggesting the pattern of DEGs in WT contribute to renal atrophy.