296 - Acute Kidney Injury unveils a rare form of Congenital Adrenal Hyperplasia
Sunday, April 27, 2025
8:30am - 10:45am HST
Publication Number: 296.3881
Marissa Lipton, Hassenfeld Children's Hospital at NYU Langone, New York, NY, United States; Olga Yeliosof, SIUH Northwell Health, Staten Island, NY, United States
Assistant Clinical Professor of Pediatrics Hassenfeld Children's Hospital at NYU Langone New York, New York, United States
Background:
Background: 17α-hydroxylase/17,20-lyase deficiency (17OHD) is a rare form of Congenital Adrenal Hyperplasia (CAH). We present a case detected during an acute kidney injury (AKI); persistence of symptoms led to further investigation. Objective:
Case Report: A 13 year old female presented with fever and sore throat. Labs showed potassium (K) 3.4 mmol/L and creatinine (Cr) 0.6 mg/dL. She had a positive PCR for streptococcus pyogenes, but ill-appearance prompted vancomycin and piperacillin-tazobactam. On hospital day (HD) #2, she had a non-oliguric AKI, with Cr 1.55 mg/dL. She was mildly hypertensive. Fractional excretion of sodium was 1.9%. Ultrasound (US) showed nephromegaly with increased cortical kidney echogenicity. Her Cr peaked at 3.2 on HD #4, but she was not discharged until HD #11 due to need for K repletion. Outpatient, her K dropped to 2.9 off supplements, despite AKI resolution. Genetic testing revealed homozygous CYP17A1 variant. She lacked sexual maturity and parents were consanguineous. Labs showed significant abnormalities in adrenal hormones (Table 1), with 46XX karyotype. A pelvic US revealed a small prepubescent uterus, without visualized ovaries. Bone age was delayed by 2 years. Design/Methods:
Discussion: AKI led to recognition of the patient’s genetic disorder, but this was not immediately obvious. Hypokalemia due to tubular wasting has been reported in both acute interstitial nephritis [1] and acute tubular necrosis [2], so significant tubular damage from combined injury was suspected. Elevated blood pressure was attributed to fluids and AKI. A rare minority (1%) of CAH is due to 17OHD [3]. Results: Patients present later than other CAH, with delayed sexual maturation, primary amenorrhea (46XX women), ambiguous genitalia (46XY men), hypertension, or hypokalemia [4]. The latter two are caused by activation of mineralocorticoid receptors, primarily due to excess 11-deoxycorticosterone, with high circulating corticosterone contributing to hypertension [5]. Androgen production is impaired, with decreased estrogen and sexual infantilism [6]. The patient’s low estrogen level with elevated gonadotropins indicate primary ovarian failure. Her bone age is delayed due to hypoestrogenism.
The treatment involves mineralocorticoid receptor antagonists, physiologic glucocorticoids, and sex steroid replacement.
Conclusion(s):
Conclusion: CAH due to 17OHD is rare. Subtle symptoms may be missed prior to puberty, after which lack of sexual maturity usually leads to diagnosis. Our patient’s AKI prompted nephrology evaluation, but persistence of hypokalemia and hypertension unveiled her underlying condition.

 photo")
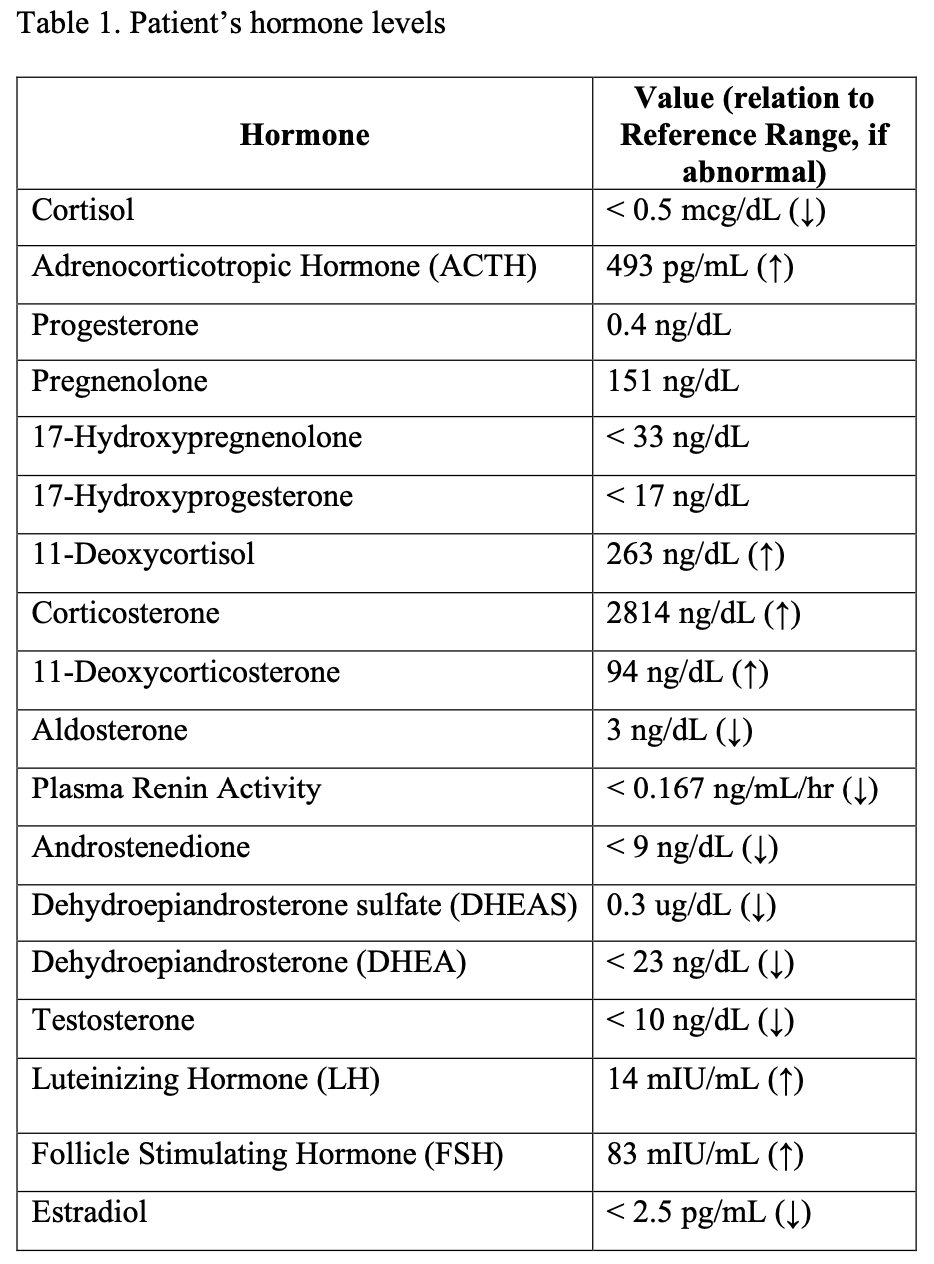 Patient’s hormone levels
Patient’s hormone levels