Postdoctoral Research Fellow Fuzhou University Affiliated Provincial Hospital Fuzhou, Fujian, China (People's Republic)
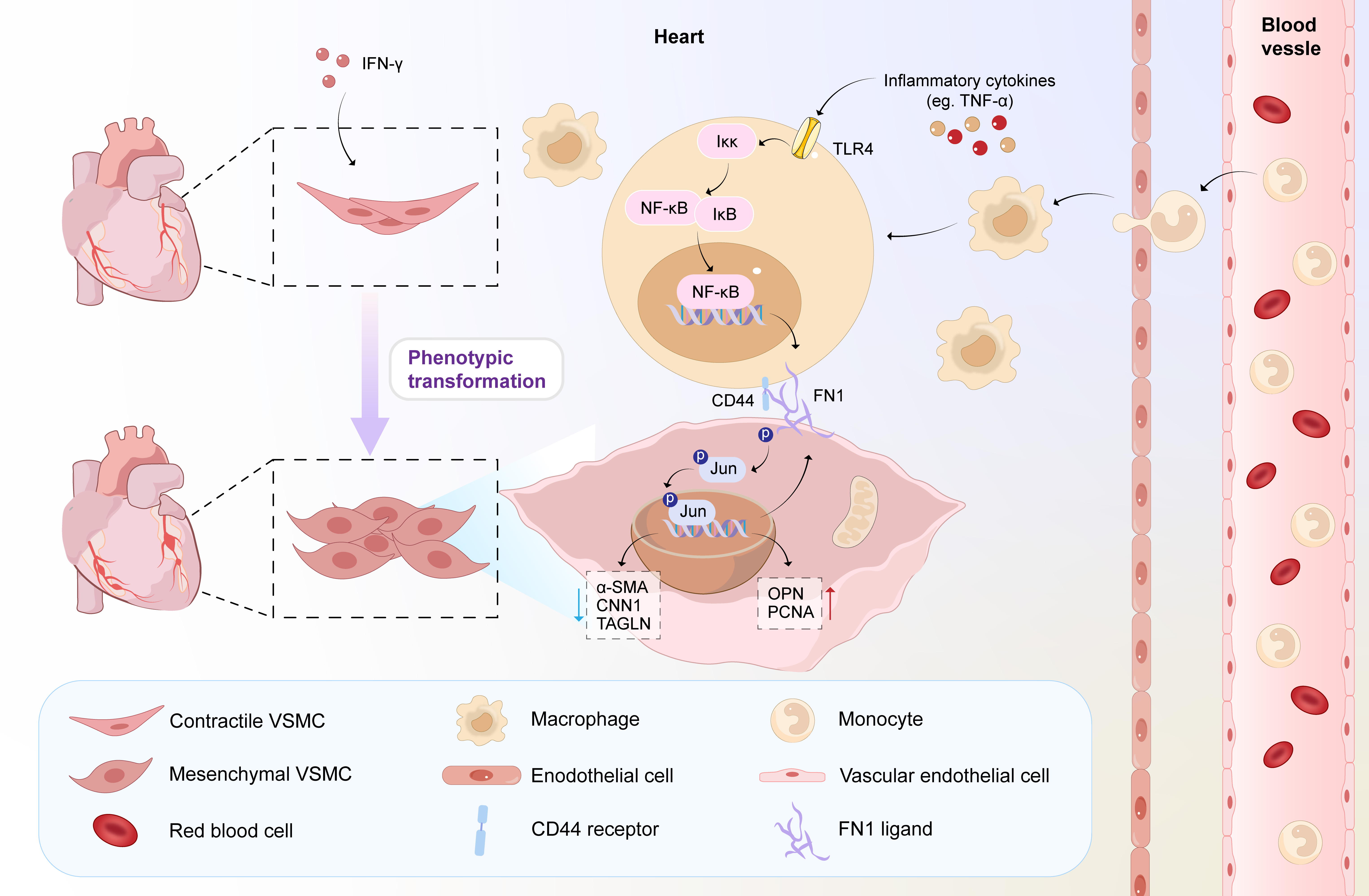
Background: Kawasaki disease (KD) is an acute vasculitis predominantly affecting children aged < 5 years, leading to coronary artery lesions if untreated. The mechanisms underlying coronary artery damage, specifically how immune cells induce vascular smooth muscle cell (VSMC) phenotypic transformation, remain unclear. We investigated the role of the JUN–FN1–CD44 signaling axis in mediating macrophage-induced VSMC dedifferentiation in KD vasculitis. Objective: This study aims to delineate the drivers behind the phenotypic switch in vascular smooth muscle cells (VSMCs) during KD progression, focusing on macrophage infiltration and transcription factor activation at single-cell resolution. Design/Methods: We analyzed coronary artery tissues from KD patients and healthy controls by integrating single-cell RNA sequencing (scRNA-Seq) data (GSE131778) and bulk RNA-Seq data (GSE64486). We performed scRNA-Seq, spatial transcriptomics, and single-cell ATAC sequencing (scATAC-Seq) on heart tissues from a Candida albicans water-soluble fraction (CAWS)-induced KD vasculitis mouse model. Results: The KD patients had significant macrophage infiltration and VSMC phenotypic switching. Motif activity analysis identified the activating protein-1 (AP-1) transcription factor complex, particularly JUN, as highly active in dedifferentiated VSMCs during acute-phase vasculitis. JUN positively regulated fibronectin (FN1) expression in VSMCs. FN1 on VSMCs facilitated the adhesion of tissue-resident CD44-positive macrophages around VSMCs, forming aggregates that promoted VSMC dedifferentiation and exacerbated vascular inflammation. In vitro, knocking down JUN in VSMCs reduced FN1 expression and prevented macrophage-induced dedifferentiation. In vivo, administering an AP-1 inhibitor, VSMC-specific FN1 knockout, or macrophage depletion using clodronate liposomes reversed VSMC phenotypic switching and reduced vascular inflammation in the mouse model.
Conclusion(s): The JUN–FN1–CD44 signaling axis is critical in macrophage-induced VSMC phenotypic transformation in KD vasculitis by enabling CD44-positive macrophages to adhere around VSMCs and form aggregates. Targeting this pathway may offer novel therapeutic strategies to prevent coronary artery lesions in KD patients.

 photo")