
Genomics/Epigenomics 2
Session: Genomics/Epigenomics 2
 photo")
Michael A. Cirelli, Jr., BA (he/him/his)
Medical Student
Mayo Clinic Alix School of Medicine
Scottsdale, Arizona, United States
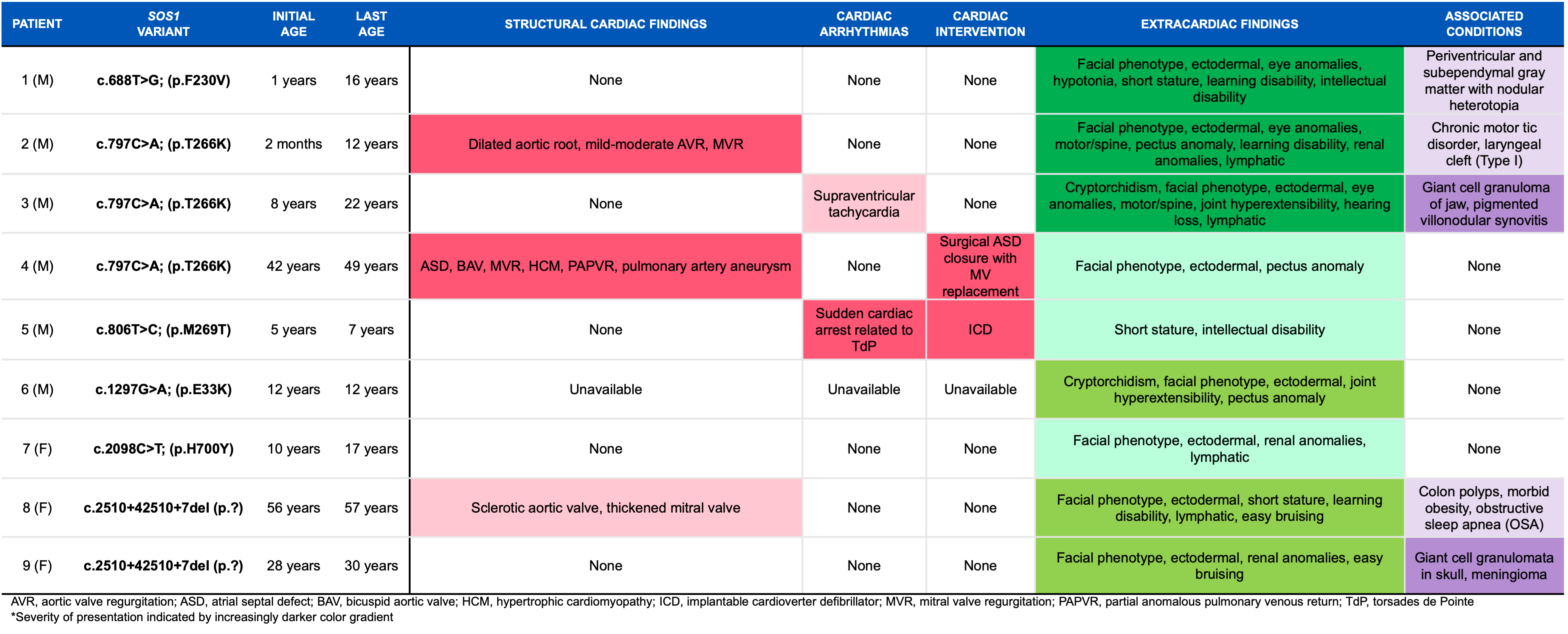 Detailed characteristics of patients with SOS1 Noonan Syndrome.
Detailed characteristics of patients with SOS1 Noonan Syndrome..png) Incidence of cancers in association with SOS1 across the literature.Detailed characteristics of patients with SOS1 Noonan Syndrome.Incidence of cancers in association with SOS1 across the literature.
Incidence of cancers in association with SOS1 across the literature.Detailed characteristics of patients with SOS1 Noonan Syndrome.Incidence of cancers in association with SOS1 across the literature.