
Genomics/Epigenomics 1
Session: Genomics/Epigenomics 1
 photo")
Tanner Ellsworth, MD, MPH (he/him/his)
Neonatal-Perinatal Medicine Fellow
University of Utah
Salt Lake City, Utah, United States
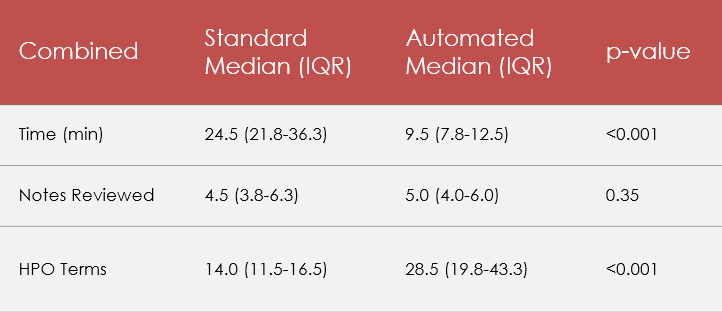 Standard vs. Pheno+ comparison, combined experience level
Standard vs. Pheno+ comparison, combined experience level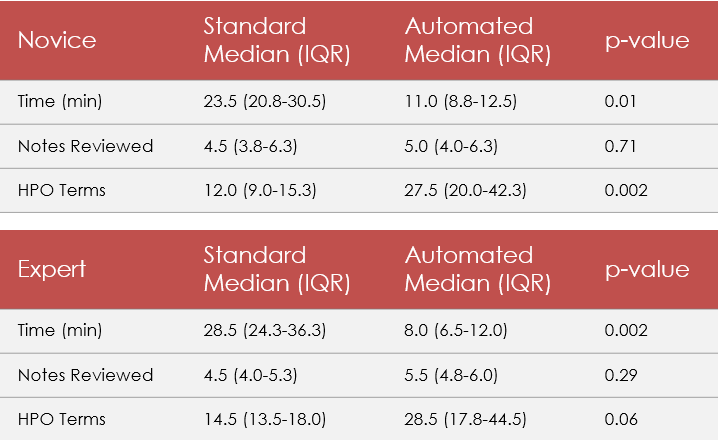 Standard vs. Pheno+ comparison, separated by clinician experience level
Standard vs. Pheno+ comparison, separated by clinician experience level