
Genomics/Epigenomics 1
Session: Genomics/Epigenomics 1
Shiyu Luo, PhD (she/her/hers)
Research assistant professor
University of Miami Leonard M. Miller School of Medicine
Miami, Florida, United States
.jpg) (A) Representative schematic of WSB2 gene (NM_018639) containing seven WD-repeats and a suppressor of cytokine signaling (SOCS) box in the C-terminus. The WSB2 variants are positioned in WD1, WD2, and SOCS box domain, respectively. (B) Common features of the three individuals carrying WSB2 homozygous variants, as indicated in grey.Key: GDD = global developmental delay; ID = intellectual disability (C) Representative magnetic resonance imaging (MRI) of individual 2 (I:2) with homozygous c.399del p.(Q134Rfs*14) at 12 months of age. (i) Sagittal T1-weighted MRI showing microcephaly, callosal hypogenesis (white arrow), tectal dysplasia (yellow arrow), and severe cerebellar hypoplasia/atrophy (dotted oval). (ii and iii) Axial T1- and T2-weighted MRI show undersulcation and white matter hypomyelination for age. (iv-vi) Coronal T2-weighted with fat suppression show small olfactory bulbs (black arrows), optic nerves (white arrows), and hippocampi (yellow arrows).
(A) Representative schematic of WSB2 gene (NM_018639) containing seven WD-repeats and a suppressor of cytokine signaling (SOCS) box in the C-terminus. The WSB2 variants are positioned in WD1, WD2, and SOCS box domain, respectively. (B) Common features of the three individuals carrying WSB2 homozygous variants, as indicated in grey.Key: GDD = global developmental delay; ID = intellectual disability (C) Representative magnetic resonance imaging (MRI) of individual 2 (I:2) with homozygous c.399del p.(Q134Rfs*14) at 12 months of age. (i) Sagittal T1-weighted MRI showing microcephaly, callosal hypogenesis (white arrow), tectal dysplasia (yellow arrow), and severe cerebellar hypoplasia/atrophy (dotted oval). (ii and iii) Axial T1- and T2-weighted MRI show undersulcation and white matter hypomyelination for age. (iv-vi) Coronal T2-weighted with fat suppression show small olfactory bulbs (black arrows), optic nerves (white arrows), and hippocampi (yellow arrows).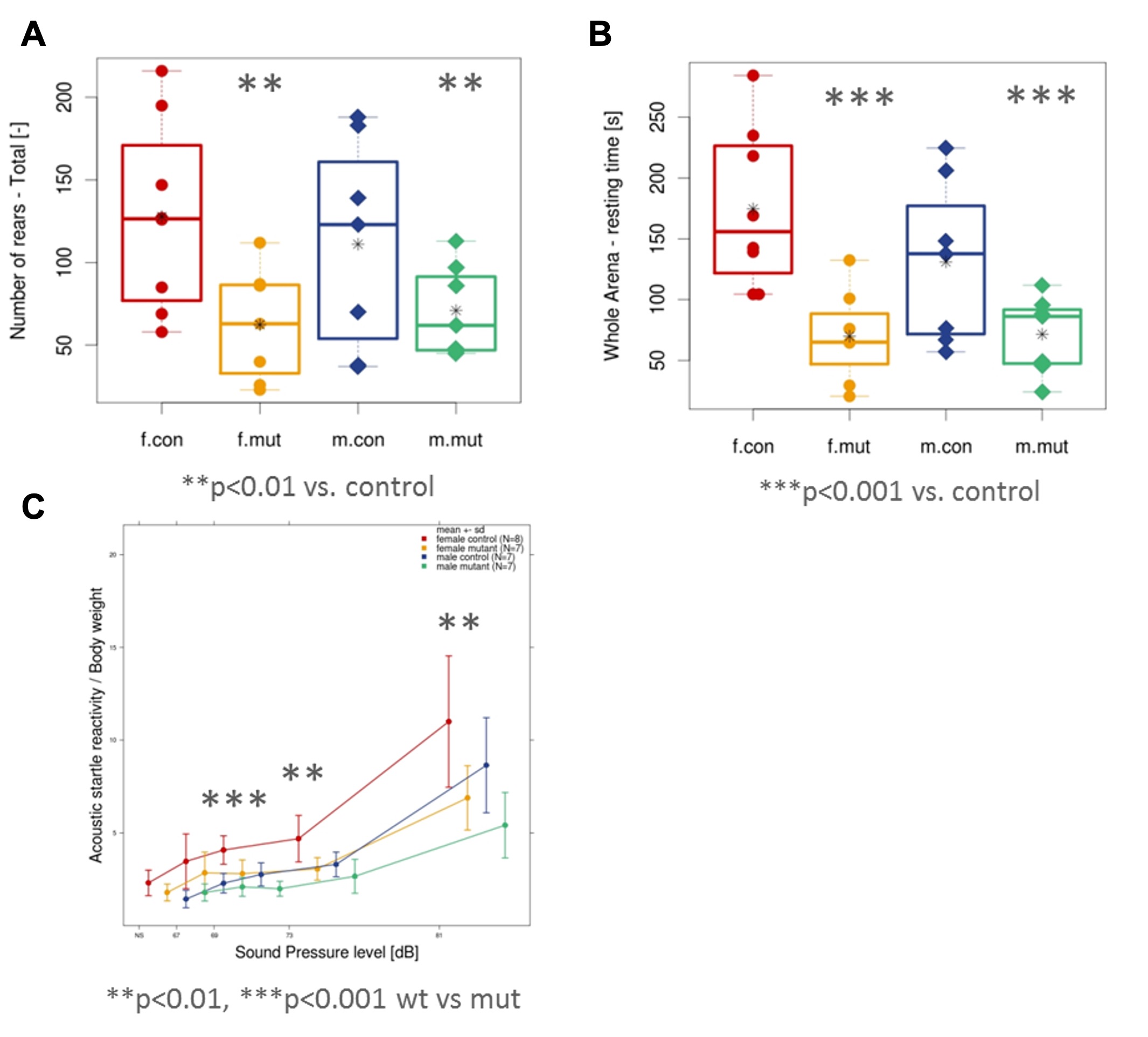 In the novel open field test, the mutant mice showed decreased rearing activity (A) and decreased resting time (B) when compared to control mice. (C) Prepulse Inhibition (PPI)/Acoustic Startle Response (ASR) testing showed decreased acoustic reactivity in the mutant mice at lower sound pressure levels.
In the novel open field test, the mutant mice showed decreased rearing activity (A) and decreased resting time (B) when compared to control mice. (C) Prepulse Inhibition (PPI)/Acoustic Startle Response (ASR) testing showed decreased acoustic reactivity in the mutant mice at lower sound pressure levels. 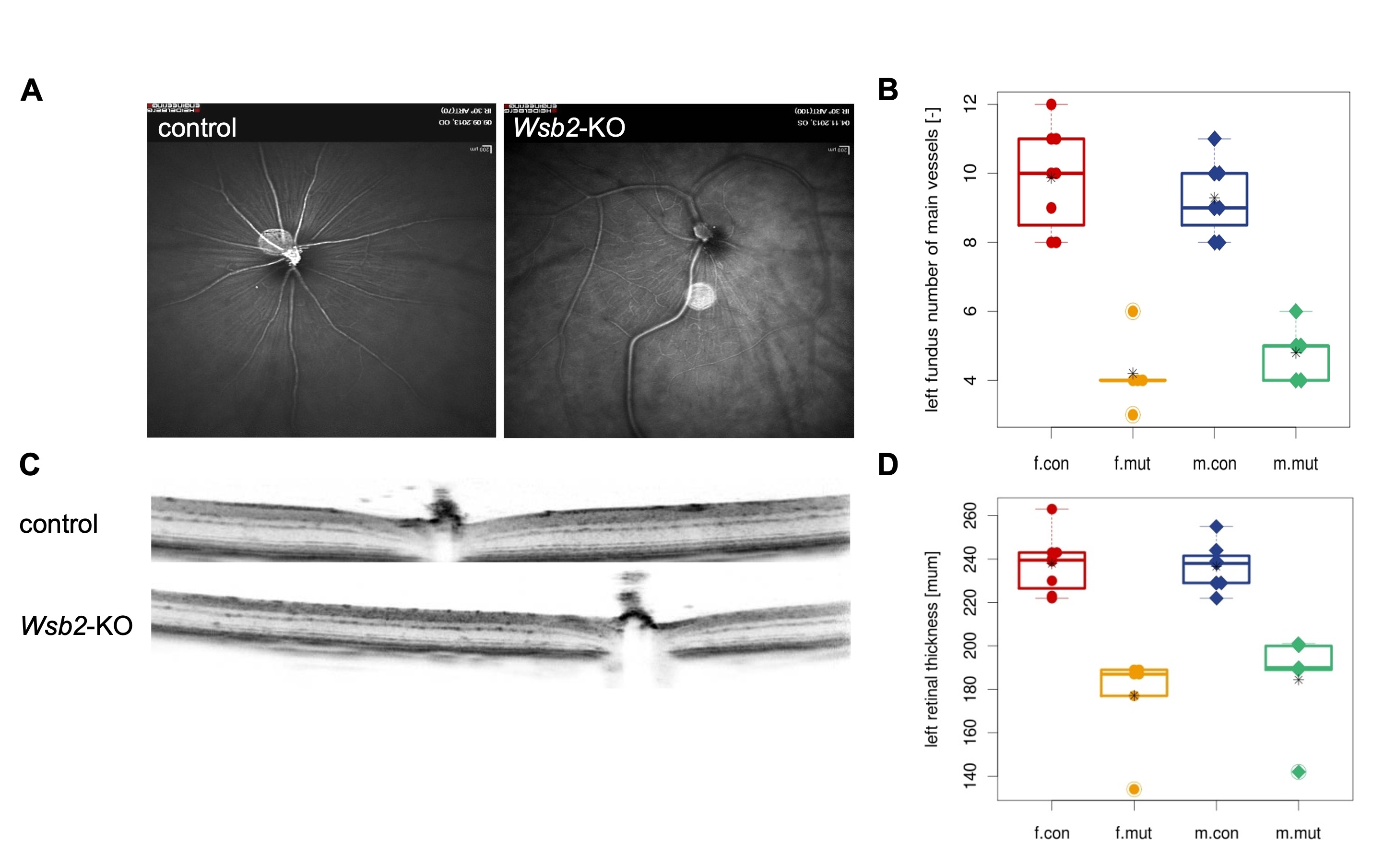 including irregular retinal blood vessel morphology (A, B) and reduced total retinal thickness (C, D) in the left eye. Controls n=15 (8 females/7 males) vs mutants n=11 (6 females/5 males). The right eye shows a similar phenotype.
including irregular retinal blood vessel morphology (A, B) and reduced total retinal thickness (C, D) in the left eye. Controls n=15 (8 females/7 males) vs mutants n=11 (6 females/5 males). The right eye shows a similar phenotype.